Using a Risk-Based Approach to Quality Management System for Medical Devices
 The risk-based approach has always underpinned the design, manufacture, control and regulation of medical devices. In most jurisdictions, medical devices are classified based on risk. Higher risk medical devices are subjected to more extensive regulatory scrutiny. This means there is greater demand for comprehensive safety data and technical documentation from manufacturers of high risk devices. Furthermore, manufacturers must demonstrate that device-associated risks have been reduced or eliminated as far as possible, regardless of risk classification. Since 2000, the ISO 14971 standard has reliably provided the guidelines, tools and methodologies specifically for medical device risk management. Written references to risk management in the ISO 13485 standard for medical device quality management system (QMS) pushes for the application of risk-based thinking beyond the product.
The risk-based approach has always underpinned the design, manufacture, control and regulation of medical devices. In most jurisdictions, medical devices are classified based on risk. Higher risk medical devices are subjected to more extensive regulatory scrutiny. This means there is greater demand for comprehensive safety data and technical documentation from manufacturers of high risk devices. Furthermore, manufacturers must demonstrate that device-associated risks have been reduced or eliminated as far as possible, regardless of risk classification. Since 2000, the ISO 14971 standard has reliably provided the guidelines, tools and methodologies specifically for medical device risk management. Written references to risk management in the ISO 13485 standard for medical device quality management system (QMS) pushes for the application of risk-based thinking beyond the product.
… apply a risk based approach to the control of the appropriate processes needed for the quality management system …
Section 4.1.2 (b), ISO 13485:2016
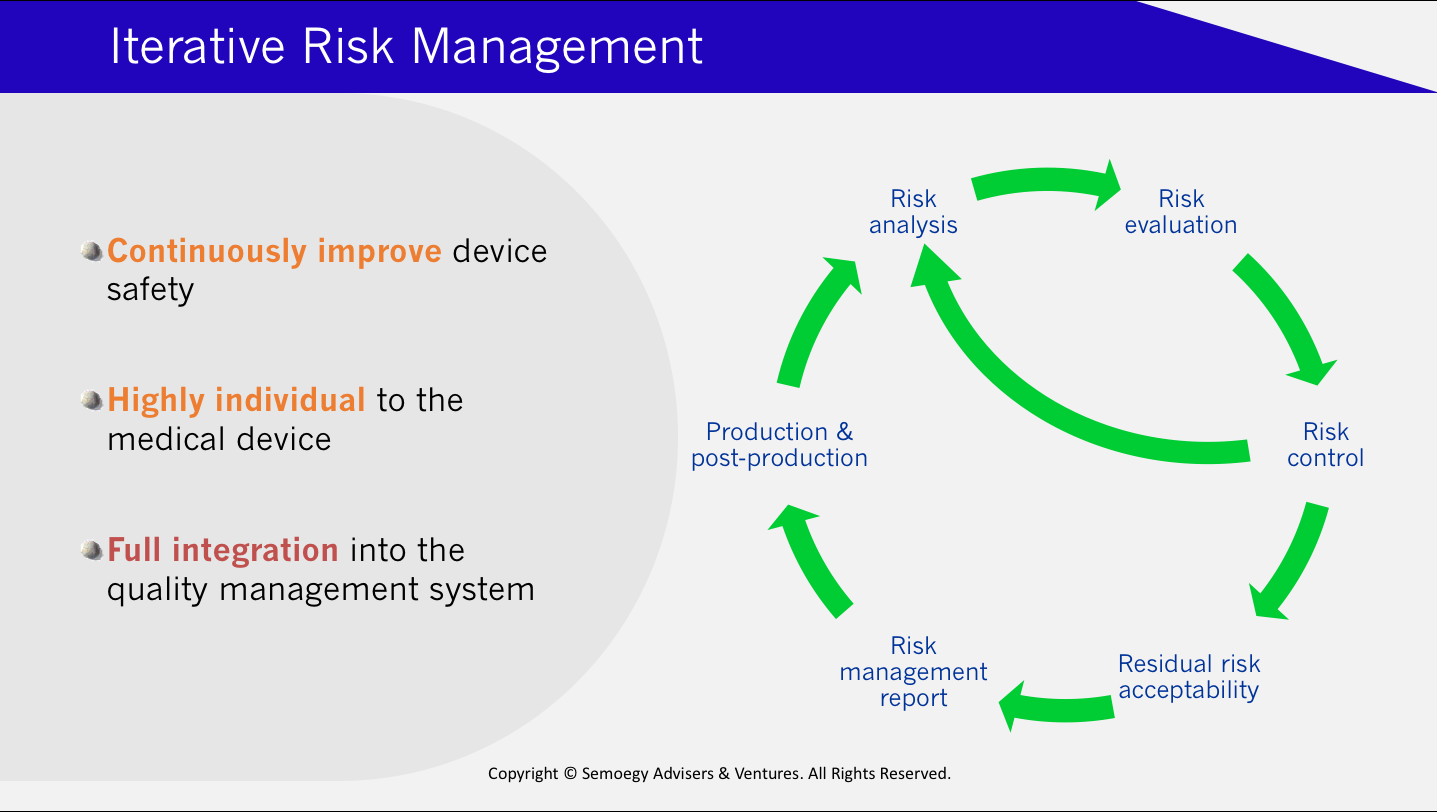
To achieve this aim, manufacturers are required to establish organisational processes and controls that incorporate risk considerations. The problem is the ISO 14971 standard puts focus on the risk management of medical devices mainly from a product perspective. Section 9 of the standard stipulates the continuation of risk management at the post-production and production stages. But it is product-focused and offers little help on how risk management can be reconciled with QMS processes. Of course, process risk management should ultimately result in risk reduction of the medical device product. Risk management at the process level safeguards product safety at every production stage, offers the benefit of real-time risk monitoring, and reduces rework.
- The Less Burdensome Pathways to US FDA Approval
- The 10 Key Chief Quality Officer Job Responsibilities for Medical Technology
Let me offer some tips how risk requirements can be effectively implemented in quality processes:
When building processes, include risk reduction goals in the process objectives
Process design begins with determining process objectives. Product technical specifications are a primary part of process objectives. At this stage, risk specifications should be included in the process objectives as well. This way, the criteria and methods to accomplish or maintain risk reduction are also incorporated into the process.
This blood line tubing example shows a separate listing of product and risk specifications. A risk specification for low microscopic defects in a blood tubing will require more control over chilling water temperatures after extrusion compared with a product specification for a clear PVC tubing.

For supplier management processes, a risk-based approach to process development may result in different procedures to manage different types of suppliers, for example, for those who are ISO 13485-certified and those who are not.
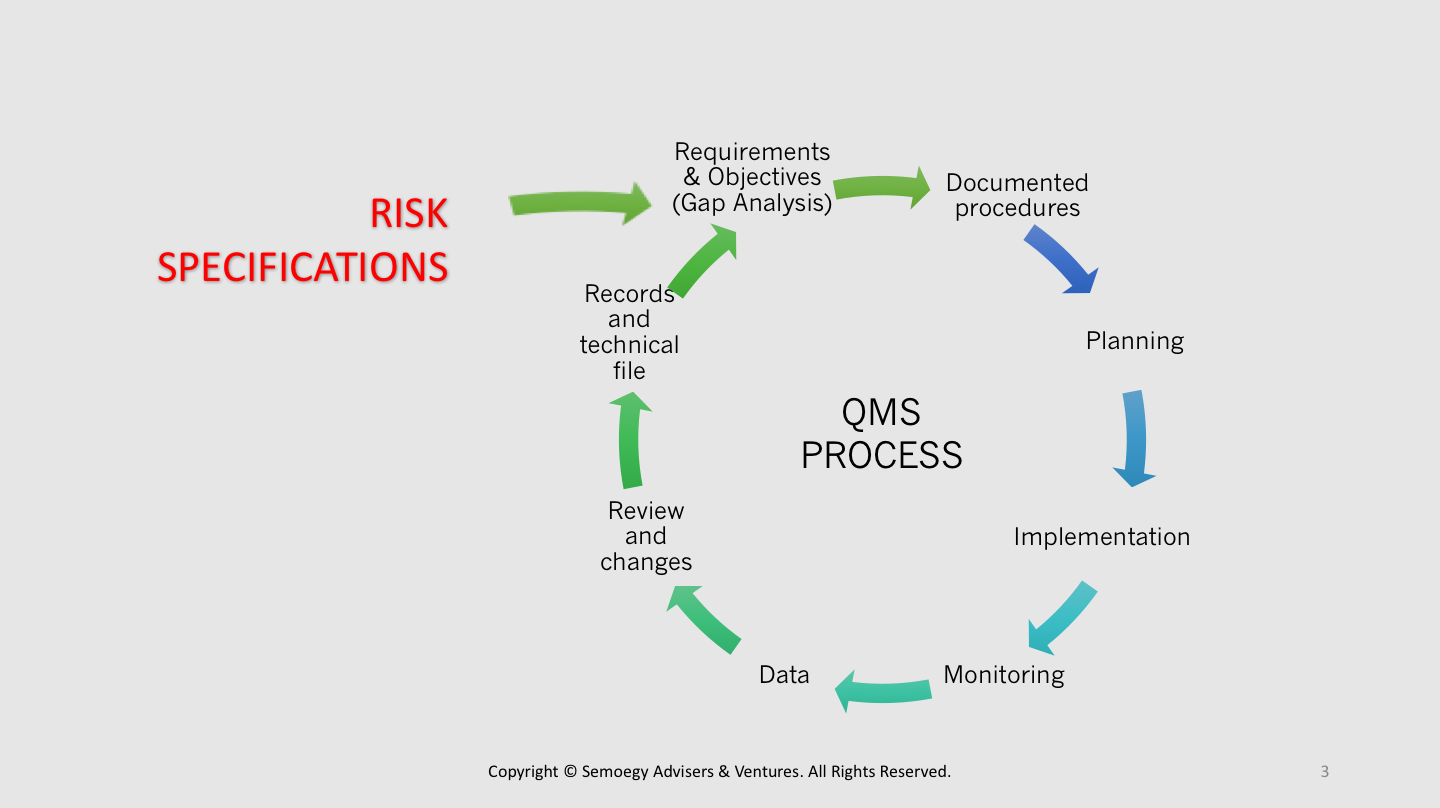
Perform risk-based gap analysis
A risk-based gap analysis is an exercise of comparing the current medical device risk levels associated with an existing process with the desired risk levels an organisation aims to achieve. Gap analysis helps a medical device organisation to keep raising the bar, identify deficiencies and seek opportunities for process improvement. When done with risk-based thinking, gap analysis enables an organisation to proactively find ways for risk reduction and control even in situations of conformity or normal situations. This is more synonymous with the risk management iteration recommended by the ISO 14971 standard.
When validating processes, include risk-based goals in process validation objectives
Process validation provides evidence that a medical devices can consistently perform in accordance with its intended use. It should be performed taking into consideration all of the conditions and environments of use. A risk-based approach to process validation should require the inclusion of all reasonably foreseeable conditions of use or misuse, in line with the ISO 14971 standard requirements. It is, therefore, vital that medical device organisations have full understanding of how medical devices are operated in real clinical settings. This includes evaluating the existing practices or deficiencies in clinical procedures and environments of various healthcare facilities that may impact medical device performance. Process validation should cover parameters that result in device performance specifications that can withstand a range of use scenarios. This is important in providing product safety assurance and documenting the performance limits of a medical device.
Use risk-based thinking to determine process data points for verification and the verification frequency
Common questions posed by medical device organisations are: (1) When to do process verification, (2) what data should be collected, and (3) at what frequency? The ISO 13485 standard requires in Section 8.2.5, the monitoring and measurement of processes (aka process verification) to demonstrate the ability of a process to achieve planned results. If risk specifications are introduced into the process objectives, then process components or parameters that result in a particular risk reduction should be verified. For example, water that has been purified through an ultrafiltration system should be tested and verified for absence of microbial contaminants. In turn, the final device should show negligible bioburden levels consistent with the use of aseptic processes. The frequency of testing may be heightened in higher risk situations when:
- A new process has been implemented
- There is a process change
- There has been a non-conformity related to the process
- There is a corrective action related to the process in response to a non-conformity
- A new medical device is produced using the process
- Re-starting a temporarily decommissioned process (for example where there is a shut-down)
- Repair and replacement work has been done related to the process
In these situations, verification may be performed more frequently and more samples taken. These data points help to establish trend lines that promote understanding and troubleshooting of the process performance. In some cases, the frequency of verification, type of data points and number of samples may be reduced over a period of time with the appropriate justification. For example, when performance or risk levels fall consistently within acceptable limits.
Implement a risk-based CAPA process
CAPA and investigation procedures that apply root-cause analysis are more likely to be in line with the ISO 14971 risk-based response to non-conformities. The ISO 14971 prescribes that the analysis of device hazards should include the study of their initiating circumstances. An appropriate tool recommended by the standard to evaluate initiating circumstances is the Fault-Tree Analysis (FTA). The FTA involves tracking of failures by back-tracing up the processes to identify the initiating events. It is an effective aid in the investigation of complaints and enables a CAPA plan that is better targeted to eliminate risks.